



Pfizerleaks: What regulators knew about the mRNA shots and journalists ought to have investigated
Insights from the Australian Therapeutic Goods Administration's evaluation documents publicly disclosed in July 2021 and available to the agency since January 2021

The Australian Government, Department of Health and Aged Care, Therapeutic Goods Administration (TGA) maintains an FOI disclosure log. “The Freedom of Information (FOI) disclosure log shows information which has been released in response to FOI requests.” Among over 40 release packages since mid-2021, a good number (but not all) regard the mRNA shots for COVID-19. For this post, I am interested in a set of nine files released on 15 July 2021, identified as FOI 2389 “Documents relating to the evaluation of the Pfizer COVID-19 vaccine”.
I cannot find information here or elsewhere on the original requester but want to thank them for their contribution to scrutinizing the mRNA vaccine approval process. I am rather unfamiliar with Australia in general, and during the pandemic as well, but I see online that the Covid Medical Network is advocating for better transparency and common-sense decision-making on the continent and has pursued other FOI requests. Another source for Pfizer documents is of course Public Health and Medical Professionals for Transparency’s listing of the US/FDA authorization files.
Disclaimer: None of the following information will be new to readers who have “done their own research”, attended “YouTube University”, and were pushed to the fringe of society as a consequence of their research. Rather, I am putting this together to illustrate that our conformist family, friends, and neighbours could have known this too, and that political opposition parties and mainstream media failed in their role to hold government and public health bureaucracy accountable for their decisions and actions.
The first document in the Australian FOI-2389 package is the “Delegate’s Overview and Request for ACV’s Advice” for the product “COMIRNATY COVID 19 vaccine” with active ingredient “BNT162b2 [mRNA]” from sponsor Pfizer Australia Pty Ltd, dated 11 January 2021. As far as I can tell, the “delegate” is a representative of the TGA who is consulting the independent Advisory Committee on Vaccines (ACV). The advice sought from the committee focuses on key characteristics of the product and the knowledge at the time. Note that the need for a benefits-risks “balance” (based on a risk/benefit analysis!?) is explicitly mentioned and the time-limited safety data and novelty of the mRNA technology are acknowledged:

As an aside, the document reports on the “overseas regulatory status” of the same product. The following list of approval dates suggests there was a race to take action (but at what cost?). Equally interesting is the language used, e.g. the well-known “emergency use authorization (EUA)” in the UK and US compared with Canada’s and Singapore’s “interim authorization”, and Switzerland’s and the EU’s “conditional authorization”:

Subsequently, the document reviews the Phase 1/2/3 trials, in particular Pfizer/BioNTech’s Study C4591001, which included about 44,000 participants globally and came up with the famous 95% vaccine efficacy. It is important to reiterate that the primary goal (“endpoint”) of the study was to reduce “confirmed COVID-19 occurring from 7 days after the second dose”. Further, “Confirmed COVID-19 cases were determined by [PCR test] and required at least 1 symptom consistent with COVID-19 disease. The symptoms included: fever, new or increased cough, new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat, diarrhoea or vomiting.”

A secondary goal was to prevent “severe COVID-19”. The initial 1:3 (prophylactic vs placebo) result was not considered statistically valid due to the very small number of occurrences. “Additional analysis conducted in all cases after first dose” yielded a more palatable 1:9 relationship … Well, aren’t these data exceptionally compliant! But as Dr. Michael Palmer put it in a recent lecture (skip to 2:41:00), “If in a clinical trial that involves 20,000 vaccinated people and 20,000 controls, you do not have enough severe disease or death in order to arrive at any kind of robust conclusion, then the only thing you have proven is that there is no emergency.”

The safety profile, shown here for the younger cohorts, is presented as solid; otherwise, I’m sure we wouldn’t have seen these products approved. However, moderate to severe adverse events were recorded and the risk/benefit profile, in particular for the younger 16-55 year age range, might not be as clear as presented to the public. Note that adverse events are reported as “reactogenicity”, which refers to expected body/immune response to the injection. For a much more critical evaluation, I refer to the Canadian Covid Care Alliance’s “More Harm Than Good” and “Stop The Shots” resources.

The discussion section in FOI-2389-01 is most informative in that it clearly and unequivocally lists the limitations of the regulator’s (and manufacturer’s) knowledge at the time of approval. This includes the lack of information on the shots’ impact on asymptomatic infection and on transmission; interactions with other vaccines; use in pregnancy; safety and efficacy in certain vulnerable groups (immunocompromised, youth, indigenous); and even their performance in a massive real-world campaign! The last bullet point seems to suggest that the immunological measure of efficacy does not even translate directly into a metric for de-facto protection...
There is more in this first document but I am already running out of space and want to verify some of these concerns on the basis of the other documents as well as scan them for any additional critical observations.
Document FOI-2389-02 is the letter from TGA to Pfizer Australia dated 24 January 2021 with the decision to “provisionally register [the] medicine” and approve the product information, which is included as an attachment. In its Section 4 Clinical Particulars, we read about a number of pertinent limitations, including that the provisional approval “has been made on the basis of short term efficacy and safety data. Continued approval depends on the evidence of longer term efficacy and safety from ongoing clinical trials and post-market assessment.” Let me reiterate: short term efficacy and safety data. Ongoing clinical trials. Post-market assessment.
The product’s indication is to provide “Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2”. Yes, prevent COVID-19! One has to wonder why these approvals were not cancelled when it became clear that the injections do not exactly prevent the disease.
While the use in children <16 years had not been approved at the time, the regulator noted the inclusion of elderly aged 65+ in the manufacturer trials. However, a note is made regarding limited data for frail elderly aged 85+: “The potential benefits of vaccination versus the potential risk and clinical impact of even relatively mild systemic adverse events in the frail elderly should be carefully assessed on a case-by-case basis.” I repeat, the potential risk and clinical impact of even relatively mild systemic adverse events in the frail elderly… Unfortunately, I doubt that vaccinators in longterm care home spend much time reading the product information and acting according to this caution.
Additional concerns known to the regulator, advisory committee, and anyone who would have sought out this information:
No interaction studies [with other medicines] have been performed.
Concomitant administration of COMIRNATY with other vaccines has not been studied.
Administration of COMIRNATY in pregnancy should only be considered when the potential benefits outweigh any potential risks for the mother and fetus.
It is unknown whether BNT162b2 [mRNA] is excreted in human milk.
The most frequent adverse reactions in participants 16 years of age and older were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia and chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually mild or moderate in intensity and resolved within a few days after vaccination.
On the basis of the August 2021 FDA approval, I have previously written (on another blog) about why the COVID jabs were still to be considered experimental, and continue to be experimental to this day. The Australian short version of the argument as of January 2021 looks like this:

This TGA document contains additional information, e.g. the ingredient list of the product, the adverse-event monitoring process, and a commitment to batch analysis with reference to past production issues. Rhetorical question: Were all vaccinees in Australia and around the world aware of the ongoing safety studies, need for risk/benefit analysis for specific recipient groups, the product’s ingredients, and possible manufacturing, transportation, and storage challenges, when they gave their informed consent to be injected with these genetics-based therapeutics?
File FOI-2389-03-1 contains information prepared for the pre-submission meeting in September 2020. This file includes a number of interesting graphics explaining the mRNA function and trials. A total of at least four vaccine candidates are mentioned: BNT162a1, BNT162b1, BNT162b2, and BNT162c2, along with multiple dosages tested between 10μg to 30 μg and up to 100μg. The BNT162b3 candidate, which was included in the fertility and developmental study I recently wrote about, is not mentioned here. From a layperson’s perspective, the following slide seems to suggest that the a, b, and c candidates use substantially different techniques: uRNA, modRNA, and saRNA.

Various sections of this file are redacted, often to hide information on the candidate cocktails, other than the final choice of BNT162b2 at 30μg. Eight “questions for discussion” at the end of a Pfizer presentation in the first part of the document are completely blacked out, including the first question regarding “safety and efficacy data”… One has to wonder who or what is being protected here.

A number of trials with varying parameters were conducted, giving the reader an idea of the enormous enterprise of “Operation Warp Speed”. One point in the copy of a BioNTech brochure speaks about the promise of anti-viral RNA “vaccines”, which may provide a glimpse at things to come:

File FOI-2389-03-2 consists primarily of an update of an amendment of the clinical protocol for the global Phase 1/2/3 trial. Towards the end, I notice in a copy of a fact sheet for healthcare providers that the US/FDA EUA is associated with an upper age limit of 85 years — I do not recall this, though maybe it was an authorization for the trial period only?
To ensure informed participant consent, healthcare providers are instructed to “communicate information from the ‘Fact Sheet for Recipients and Caregivers’ (and provide a copy of the Fact Sheet), to the recipient or their caregiver, including the following” (my emphasis):
FDA has authorized the emergency use of the Pfizer-BioNTech COVID-19 Vaccine, which is not an FDA-approved vaccine.
The individual or caregiver has the choice to accept or refuse the Pfizer-BioNTech COVID-19 Vaccine.
All potential risks and benefits of the Pfizer-BioNTech COVID-19 Vaccine are
not yet known.Information about available alternative vaccines and the risks and benefits of
those alternatives.
No word on alternative prophylaxis nor treatment options. And was any of the above information later provided at COVID-19 mass vaccination clinics and drugstore drop-ins?
Still on the same fact sheet, we read that “each of the following requirements must be met”, including:

That makes it pretty obvious that VAERS is indeed a suitable database to study the collateral damage from the shots in the US, despite all talk about the supposedly voluntary reporting and the quality issues with basic elements such as the batch ID. Here, serious adverse events are defined quite tightly starting with death. Yet, they include the relatively broad category of “persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions.”

Lastly, the recipient/caregiver fact sheet includes the revealing note that “As with any vaccine, the Pfizer-BioNTech COVID-19 Vaccine will not protect all persons who are vaccinated.” Additionally, pregnant and breastfeeding mothers were advised (at the time) not to get the shots. Certainly, mainstream investigative journalists and science writers have carefully examined the updated risk (safety) vs benefit (efficacy) profile of the shots as their use in pregnant women is now common-place?

File FOI-2389-04 contains the meeting minutes of the ACV regarding the BNT162b2 authorization application. It includes a lot of the same information already discussed above. Of interest is another listing of the limitations of the data available to support the decision to be made (“Delegate’s summary of issues”), including (no emphases although I want to highlight every single word in this list!):
Safety follow up is currently limited to median two months post Dose 2.
The duration of immune response and vaccine protection is not currently known.
Vaccine efficacy against asymptomatic infection and viral transmission are not yet
known.The data in immunocompromised individuals are very limited.
Lack of data in paediatric subjects, pregnant women, and lactating mothers.
The agency representative then puts a number of related questions before the independent scientists who are members of the committee:

The committee’s discussion revolves around SARS-CoV-2 epidemiology, specifically in Australia; international approvals; product quality with a mention of concerns regarding doses-per-vial, the “presence of truncated/modified forms of mRNA at higher concentrations than in clinical trial” noted by the European regulator (discussed primarily as a concern regarding the efficacy of the injections); “increased DNA contamination” as a potential cause for increase side effects; and lastly monitoring the necessary freezing temperatures “throughout the supply chain”.
In the discussion of efficacy, the ACV notes a long list of limitations in excess of what the agency brought to the table already (see above):
Protection from long-term complications secondary to SARS-CoV-2 infection, and effect on mortality, are not yet known
Lack of data in children under 12 years (and limited data for those 12-15 years which was not reviewed in this submission), pregnant women, and lactating mothers
No data on concurrent administration with other vaccines
No data on mixed schedules with other potential COVID-19 vaccines
An immunological correlate of protection is not established
No data are available in the Aboriginal and Torres Strait Islander population
The sponsor had not provided immunogenicity subgroup analysis by subdividing the age groups, such as 75-85 year olds
There were few participants with renal failure as a co-morbidity, or other groups who may respond poorly to vaccines
There are limited data to inform the duration of protection provided by this vaccine.
In terms of safety, the ACV notes a number of positive trial outcomes. They consider the trials powered enough to identify safety issues up to 1:5,000. Note that this is the order of magnitude currently assumed for myocarditis incidence after vaccination, so this may or may not have been detected, while it is now widely acknowledged as a valid safety concern. The committee also notes four occurrences of Bell’s Palsy in the vaccine arm of the C4591001 study and discusses reports of anaphylaxis ascribed to the polyethylene glycol contained in the shots.
The limitations/requirements regarding safety that were recorded by the ACV are as follows:
No studies of complement activation and cytokine stimulation.
Safety concerns to be addressed in ongoing and planned pharmacovigilance activities:
Anaphylaxis
Vaccine-associated enhanced disease (VAED) including vaccine-associated
enhanced respiratory disease (VAERD)Use in patients with co-morbidities, including frail patients with co-morbidities (e.g., chronic obstructive pulmonary disease, diabetes, chronic neurological disease, cardiovascular disorders)
Use in immunocompromised patients, and patients with autoimmune or
inflammatory disordersUse in pregnancy and while breast feeding
Interaction with other vaccines
Long term safety data.
In terms of post-marketing risk management, the committee reviewed the studies planned and under way; discussed enhancement to active and spontaneous (passive) surveillance programs for vaccine adverse events; emphasized “the importance of determining the background rate of syndromes and diseases that may be regarded as potential adverse events given the likely widespread use of this vaccine”; and proposed a (short) list of adverse events of special interest (AESI), a concept used in Canada and elsewhere too.
The committee ultimately supports the provisional registration of the product. They advise to remove a reference to “an officially declared pandemic” from the drug’s indication; I am not sure why this was proposed by Pfizer/TGA but it does remind me of the scrutiny after the 2009 swine flu “pandemic” that indicated that the premature declaration of a pandemic by the WHO only enabled a global overreaction. So, is the reference removed here to ensure that the vaccination campaign can continue independently of a possible end of the “pandemic”?
In terms of the question about the very limited 2-month followup, “The ACV advised that it is unlikely for vaccine-related adverse events to occur more than 2 months after vaccination based on available data. However, there is limited information on the use of mRNA vaccine in humans, which underpins the need for post market vaccine safety surveillance.” That sounds rather contradictory, if not concerning.
The committee also recommends some tightening of the pharmacovigilance plans and product information such as highlighting the uncommon “do not shake” instructions for the vaccine vials and making “The minimum 15 minute observation period following administration … mandatory, not merely recommended.”
Document FOI-2389-05-1 is the “Clinical Evaluation Report” of BNT162b2 dated 8 January 2021. In the background section, I notice what I believe is largely misinformation regarding the transmission of SARS-CoV-2, namely asymptomatic spread (not sure about the latest on this), transmission via droplets (isn’t it aerosols?), and surface infection (debunked since at least April 2020). This is surely not a great start to evaluating the benefits-risks balance for the mRNA shots in an unbiased manner.

The remainder of this report focuses on the immunogenicity (triggering an immune system reaction) results from the trials. The contents is too technical for me to comment much, but I want to share a chart that shows the concentrations of IgG antibodies in the participants’ blood at the times of doses 1 (Day 1) and 2 (D21), and seven (D28), 14 (D35), and 31 days (D52) after dose 2. I can’t help but notice the decline from the peak starting one week after dose 2. The D35 and D52 declines are not commented by the evaluator despite the stated concern quoted above that the “duration of immune response and vaccine protection is not currently known”. And indeed, we have since observed rapidly waning vaccine efficacy within months if not weeks after vaccination!
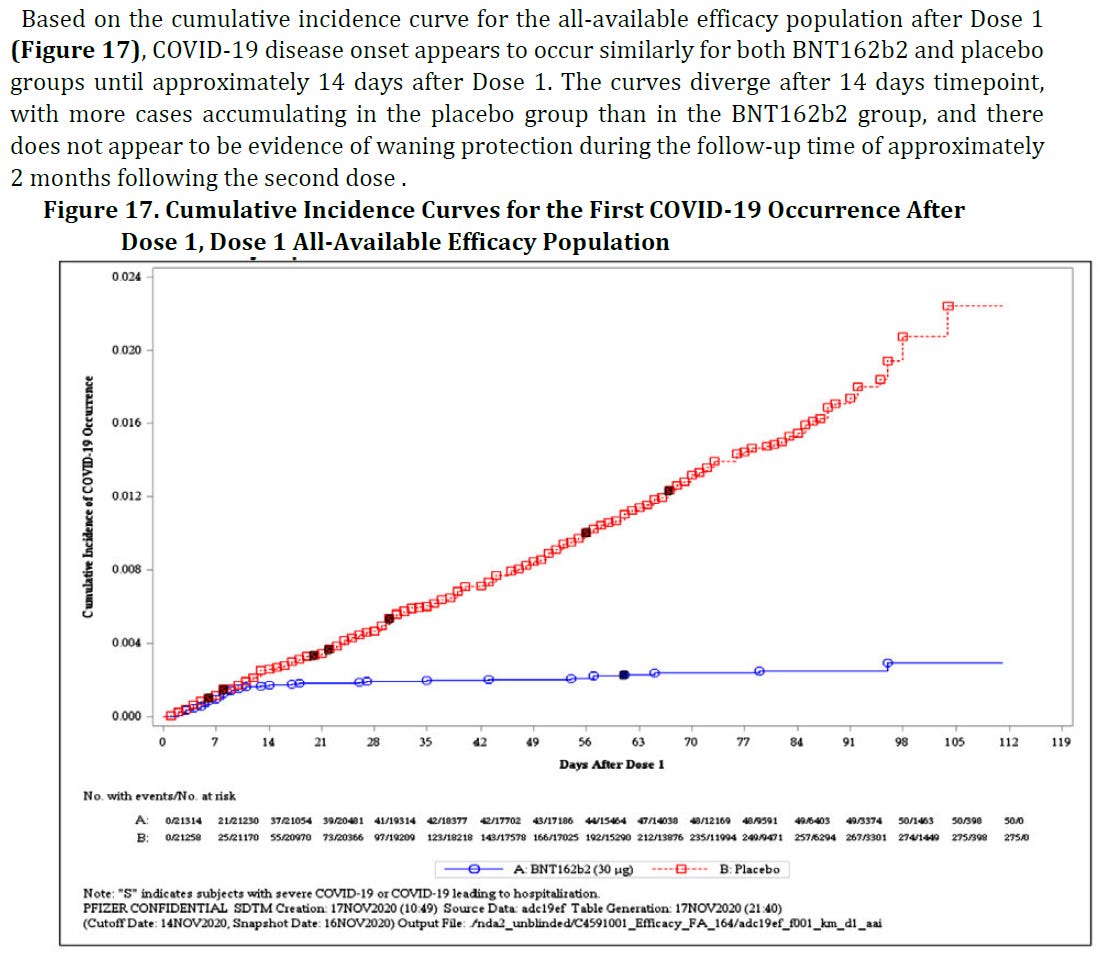
Given the data received from Pfizer/BioNTech, including another chart that shows continuously growing COVID-19 incidence in the placebo group contrasted with an all-but-flat curve for the vaccinated group, the regulator could be excused for concluding that the injections were highly effective at the time. Except that in discussing the evaluation by the European Medicines Agency during the above-mentioned lecture, Dr. Palmer puts his finger on a discrepancy between the last two charts that I included: The cumulative incidence curves start to diverge sharply just 7-10 days after dose 1 (!) while the antibody titers in the previous barchart grow much more slowly over a period of several weeks. One possible explanation of the earlier effect is that the vaccinated participants have been tested less frequently than those in the placebo arm, which would imply fraud and is just a hypothesis at this point.

As already noted, the secondary endpoint of Pfizer’s large Phase 1/2/3 trial, protection from severe COVID-19, could not be demonstrated. This becomes more clear here, where the confidence interval includes negative efficacy for individual time frames, and for the 1:9 ratio for severe illness at any time after dose 1, the 89% efficacy is bound by a confidence interval that reaches as low as 20%, which would not meet the threshold for vaccine approval (e.g. 50% risk reduction, or 30% as the Australian documents seem to suggest). Also, as is well-known by now: “The phase 2/3 study data does not provide any information on the effect of the vaccine in preventing asymptomatic infection or effectiveness against transmission of SARS-CoV-2.”
The remainder of FOI-2389-05-1 along with FOI-2389-05-2 assess the clinical safety data but I will skip this information here entirely.
The document identified as FOI-2389-06 contains the “Nonclinical Evaluation Report”, marked as last revised on 15 January 2021. This report deals primarily with immunogenicity, pharmacokinetics (distribution), and toxicity results of animal studies completed in mice and monkeys. I will limit my review to the summary section at the beginning of the port, which includes a number of noteworthy points (my emphases):
BNT162b2 (V9) was found to be immunogenic in non-clinical studies in mice, rats and rhesus macaques. BNT162b2 (V9) induced humoral and cellular immune responses in mice and monkeys. However, antibodies and T cells in monkeys declined quickly after 5 weeks after the second dose of BNT162b2 (V9) raising long term immunity concerns.
The vaccine protected monkeys from infection when challenged 55 days after the 2nd vaccine dose based on viral RNA load and radiographic lung lesions. The vaccine dose in monkeys (100 μg) was higher than the proposed clinical dose of 30 μg.
Almost (sic!) similar microscopic lung inflammation was observed in both challenged control and immunised animals after the peak of infection (Days 7/8). Rhesus macaques do not show clinical signs and generally develop only mild lung pathology from SARS-CoV-2 infection. There were no studies on protection of older animals from SARS-CoV-2 infection or duration of protection after immunisation. The animal studies were of short term; long term immunity was not assessed. The sponsor indicated that long term immunity would be addressed by human data.
There are no distribution and degradation data on the S antigen-encoding mRNA. A whole body imaging study with a surrogate, luciferase expressing mRNA indicate that the vaccine LNP formulation is expected to deliver the mRNA effectively in vivo, the mRNA and translated antigen protein are mainly localised at the injection site, distributed in liver and likely draining lymph nodes, and nearly completely degraded in 9 days.
To recap, we see concerns regarding the duration of protection (waning efficacy); lack of studies of long-term immunity; the short-term protection in monkeys was from a significantly higher dosage than is being used in humans; injected animals and control group suffered similar lung inflammation; mRNA may or may not be spread throughout the body; older animals were not tested; and human guinea pigs were going to be used (and continue to be used) to assess long-term immunity with the results currently on full display in the real world.
The distribution & degradation point may be worth another comment. The summary mentions only injection site, liver, and lymph nodes yet the report includes Table 4-2, which shows distribution to various other organs, notably the ovaries in female animals. That dataset only goes to 48 hours after injection; it is unclear to me where the claim arises that after nine days, the vaccine is “nearly completely degraded”. On 17 March 2022, Dr. Jessica Rose wrote about a document from the Pfizer release regarding study R-20-0072 in Wistar rats. That document contains a table for biodistribution results with columns going up to nine days, yet the actual data were entirely redacted. Without disclosing any details, the British Medicines
and Healthcare products Regulatory Agency (page 17) claims regarding that same study R-20-0072 that “Luciferase expression was identified at the injection site at 6 hours after injection and diminished to near baseline levels by day 9.”
The last document of the FOI-2389 set is the Australia-specific annex to Pfizer’s risk management plan (RMP). An RMP for drug approval describes the measures taken to prevent or mitigate any risks identified in the approval process through additional research studies and pharmacovigilance, here: vaccine adverse effect surveillance.

The document’s Section 2.2. Summary of the Safety Concerns consists primarily of a short tabulation representing a range of concerns from the European Union’s RMP. These seem identical to the ACV’s list of open safety concerns, which was included earlier in this post, with a bit of reordering. The only significant difference I can find is that the ACV noted concerns with the “Use in patients with co-morbidities, including frail patients”, while the EU limits this concern to just “Use in frail patients with co-morbidities”. No additional safety concerns are noted for Australia as compared to the EU.
After outlining routine pharmacovigilance processes in Australia, the report turns to the ongoing clinical studies, in particular C4591001. The statement that “The primary efficacy analysis was efficacy against COVID-19 at least 14 days after the last dose of vaccine in participants without evidence of prior SARS-CoV-2 infection at baseline” is inconsistent with the above-mentioned primary endpoint for a 7-day period and the inclusion of those with and without prior infection. However, the current record for the study in the US National Library of Medicine includes both variants for multiple outcome measures.
Of note too is the deadline for the final clinical study report for C4591001, which is stated for the end of the year 2023. Unfortunately, the trial was compromised in a major way when the placebo group was unblinded in early 2021. As the Canadian Covid Care Alliance (slide 9) puts it, “It’s no longer a randomized control trial, as [the] control group is gone” and “The long term safety data that was supposed to be assessed [in May 2023] is no longer possible to ascertain as the placebo group crossed over two years previously.”

My last observation concerns the handling of three of the eight remaining safety concerns. There is no routine monitoring planned nor additional risk minimization. In one instance, “local product information” will be adapted, meaning that a warning will be added to the product flyer. The only measure taken to address concerns for people with autoimmune or inflammatory disorders, multi-vaccine interaction, and long-term safety data are additional studies, including the completion of the compromised C4591001.
I am looking at this in the context of vaccination mandates that have since come down the pipeline and the highly restrictive handling of medical exemptions, when in fact all the safety concerns mentioned in the FOI documents should have provided ample grounds not just for exemptions but for the free and informed choice not to take the jab.